5 mg plendil purchase fast deliverySex-relatedeffects the genetic specification of sexual differentiation is described in Chapter forty three hypertension 4011 cheap 5 mg plendil with amex. The Y carries a small variety of genes involved with improvement and maturation of masculine options and in addition sections homologous with components of the X blood pressure medication migraines plendil 10 mg buy mastercard. Y-linked traits (of which there are very few) are therefore confined to males, but X-linked can show a criss-cross pattern from fathers to daughters, mothers to sons down the generations. The most significant facet of sex-related inheritance issues X-linked recessive alleles, of which there are numerous. Mitochondrialinheritance the items of inheritance corresponding to Mendel described are carried on the autosomes (non-sex chromosomes), which exist in homologous pairs. In addition there are multiple copies of a much smaller genome in nearly every cell of the human physique, which resides within the tiny subcellular organelles referred to as mitochondria (see Chapter 12). The mode of inheritance of mitochondria derives from the mechanism of fertilization. They carry little else but a nucleus, a structure that assists penetration of the ovum and a tail powered by a battery of mitochondria. The latter are nevertheless shed earlier than the sperm nucleus enters the ovum and so make no contribution to the mitochondrial population of the zygote. By contrast the ovum is very large and loaded with nutrients and plenty of copies of the subcellular organelles of somatic physique cells (see Chapter 14). All the genes carried within the mitochondrial genome are subsequently passed on solely by females, and equally to offspring of both sexes. Mitochondrial inheritance is subsequently entirely from moms, to offspring of each sexes. In achondroplasia, a type of short-limbed dwarfism, homozygotes for the dominant achondroplasia allele are so severely affected that they die in utero. The consequence is that the reside offspring of heterozygous achondroplastic companions happen in the ratio of two affected not three, to every unaffected recessive homozygote (see Chapter 5). The expression of many genes is modified by alleles of other genes as well as by environmental components. Many genetic conditions therefore show variable expressivity, complicated the concept of easy dominance. In some circumstances an apparently dominant allele could appear to skip a era because its expression in a single carrier has been negated by other factors. For instance, when a website on the long arm of the maternally derived chromosome 15 has been deleted it offers rise to Angelman syndrome within the offspring. Children with this condition show jerky actions and are severely mentally handicapped. When the equal site is deleted from the paternally derived chromosome 15, the child is affected in a really completely different way. These kids have Prader�Willi syndrome, characterized by features that embody compulsive consumption of meals, weight problems and a lesser degree of psychological handicap. He was actually unaware of the importance of chromosomes in that regard and the traits he described confirmed independent assortment with each other. Around 20 human genetic ailments develop with rising severity in consecutive generations, or make their appearance in progressively younger sufferers. Meioticdrive Heterozygotes produce two sorts of gametes, carrying various alleles at that locus and the proportions of the offspring described by Mendel point out equal transmission of those alternate options. Rarely one allele is transmitted at greater frequency than the opposite, a phenomenon known as meiotic drive. In addition, environmental factors modify phenotypes, further blurring genetically based mostly distinctions (see Chapters 50 and 51). Examples of straightforward dominant and recessive conditions of nice medical significance are familial hypercholesterolaemia (Chapters 5 and 6) and cystic fibrosis (Chapter 6). Some alleles which are particularly important in medication are revealed only when people are exposed to unusual chemicals. These guidelines apply solely to circumstances of full penetrance and where no novel mutation has arisen. Direct transmission through three generations is virtually diagnostic of a dominant. Dominant illness allele homozygotes are extremely rare and with many illness alleles homozygosity is deadly or causes a extra pronounced or extreme phenotype. Matings between heterozygotes could contain inbreeding (see Chapter 5), or occur when sufferers have met as a consequence of their disability. Example the primary situation in people for which the mode of inheritance was elucidated was brachydactyly, characterised by abnormally short phalanges. Unrelated marriage companions are subsequently often recessive homozygotes (bb) and the mating can be represented: Bb � bb Bb,bb 1: 1 Dominant illness alleles are stored at low frequency since their carriers are less match than regular homozygotes. For instance: 1 For the offspring of a heterozygote and a traditional homozygote (Bb � bb 1 Bb; 1 bb), risk of B� = 1/2, or 50%. Caf�-au-lait patches, dermal fibromas, macrocephaly, scoliosis, learning difficulties. Osteogenesisimperfecta Highly variable, with multiple fractures and lens deformity. Myotonicdystrophy Progressive muscle weakness with inability to chill out muscle tone normally, cataracts, cardiac conduction defects, hypogonadism. Death may end up from concentration of haem within the liver, following induction of haem-containing Cytochrome P450 proteins. Estimation of mutation price the frequency of dominant ailments in households with no prior circumstances can be used to estimate the natural frequency of latest point mutations (see Chapter 26). This varies broadly between genes, however averages about one mutational occasion in any particular gene per 500 000 zygotes. Almost all point mutations come up in sperm, each containing, on the newest estimates, 20�25 000 genes (see Chapter 19). There are subsequently perhaps 25 000 mutations per 500 000 sperm, so we are in a position to anticipate round 5% of viable sperm (and babies) to carry a new genetic mutation. However, solely a minority of these occurs inside genes that produce clinically significant results, or would behave as dominant traits. Halothanesensitivity,malignanthyperthermia (geneticallyheterogeneous) One in 10 000 patients can die in excessive fever when given the anaesthetic halothane, especially in combination with succinylcholine. One such is debrisoquinehydroxylase, involved in the metabolism of the antihypertensive debrisoquine and other medicine. Skeleton Affected people have joint laxity, a span:top ratio larger than 1. There are unusually lengthy, slender limbs and fingers, pectus excavatum (hollow chest), pectus carinatum (pigeon chest) and scoliosis that may trigger cardiac and respiratory issues. Heart Most sufferers develop prolapse of the mitral valve, its cusps protruding into the left atrium, permitting leakage back into the left ventricle, enlargement of which might end up in congestive heart failure. More critical is aneurysm (widening) of the ascending aorta in 90% of patients, leading to rupture during exercise or being pregnant.
Diseases - Sutton disease II
- Ptosis coloboma trigonocephaly
- Trichothiodystrophy
- Meningomyelocele
- Epidermoid carcinoma
- Hypoplasia hepatic ductular
- Glyceraldehyde-3-phosphate dehydrogenase deficiency
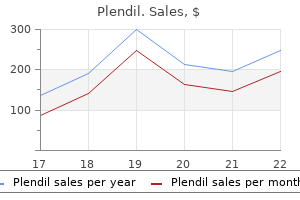
Plendil 2.5 mg purchase lineSpinal cord compression because of pulse pressure 120 effective plendil 5 mg atlantoaxial instability is a preventable complication fetal arrhythmia 36 weeks plendil 10 mg discount mastercard. Spranger J, Bierbaum B, Herrmann J: Heterogeneity of Dyggve-Melchior-Clausen dwarfism, Hum Genet 33:279, 1976. Dimitrov A, et al: the gene liable for DyggveMelchior-Clausen syndrome encodes a novel peripheral membrane protein dynamically associated with the Golgi equipment, Hum Mol Genet 18:440, 2009. Spondylometaphyseal dysplasia comprises a group of issues in which the spine and metaphyses of the tubular bones are affected. At least seven sorts have been classified based mostly on minor radiographic differences and mode of transmission. Growth deficiency, especially of trunk, with onset from 1 to 4 years of age; adult height, 129. Short neck and trunk with dorsal kyphosis; generalized platyspondyly with anterior narrowing in thoracolumbar region on lateral roentgenograms; on anteroposterior view, vertebral bodies prolong more laterally to pedicles producing an "open-staircase" appearance; odontoid hypoplasia. Irregular rachitic-like metaphyses, particularly the proximal femur with very short femoral necks; brief, stocky hands; hypoplastic carpal bones with late ossification (delayed bone age). A noticeably waddling gait with limitation of joint mobility turns into apparent at 15 to 20 months and References Kozlowski K, Maroteaux P, Spranger J: La dysostose spondylo-m�taphysaire, Presse Med 75:2769, 1967. Metatropic derives from the Greek word metatropos, which implies "changing patterns" and refers to the change in physique proportions from brief limb/long trunk to quick trunk/long limb as kyphoscoliosis turns into more progressive. There are multiple causes of the respiratory difficulties, including abnormalities of the thorax, irregular vocal cords with arytenoid fusion, and laryngotracheomalacia. The major reason for demise is cardiorespiratory failure attributable to kyphoscoliosis and the slim thorax. All cases ranging from perinatal lethality to gentle forms are as a result of mutations of the same gene. Birth weight regular; birth length higher than 97th percentile; trunk, initially long relative to the limbs, becomes progressively brief with the development of kyphoscoliosis, leading to short-trunk dwarfism. Early platyspondyly with progressive kyphosis and scoliosis in infancy to early childhood; odontoid hypoplasia; delayed ossification/hypoplasia of the anterior portion of the primary cervical vertebra, C1-C2 subluxation; narrow thorax with short ribs; quick limbs with metaphyseal flaring and epiphyseal irregularity with hyperplastic trochanters; prominent joints with restricted mobility at knee and hip but increased extensibility of finger joints; irregular and squared-off calcaneal bones and precocious calcification of the hyoid and cricoid cartilage; erratic areas of microcalcifications in vertebral our bodies and epiphyses; hypoplasia of basilar pelvis with horizontal acetabula, quick deep sacroiliac notch, and squared iliac wings. The trunk, originally long, becomes extremely short secondary to quickly progressing kyphoscoliosis. Odontoid hypoplasia with C1-C2 subluxation can result in wire compression, quadriplegia, and typically dying. Cervical References Fleury J, et al: Un cas singulier de dystrophie ost�ochondrale cong�nitale (nanisme m�tatropique de Maroteaux), Ann Pediatr (Paris) 13:453, 1966. Beck M, et al: Heterogeneity of metatropic dysplasia, Eur J Pediatr 140:231, 1983. Shohat M, et al: Odontoid hypoplasia with vertebral cervical subluxation and ventriculomegaly in metatropic dysplasia, J Pediatr 114:239, 1989. Kannu P, et al: Metatrophic dysplasia: Clinical and radiographic findings in eleven patients demonstrating long-term natural historical past, Am Med Genet 143:2512, 2007. Genevieve D, et al: Revisiting metatropic dysplasia: Presentation of a sequence of 19 novel patients and review of the literature, Am J Med Genet 146:992, 2008. Note the midface hypoplasia, large joints, short limbs, relatively giant toes and palms, and congenital scoliosis. The time period geleophysic (geleos, which means "joyful" and physis, meaning "nature") refers to the happy-natured facial appearance typical of this disorder. All the survivors have had cardiac involvement, though gentle and asymptomatic in some. Tracheal narrowing appears to significantly affect consequence, as does the extent of the cardiac involvement. Short stature predominantly of postnatal onset with regular upper/lower segment ratio, span is decreased, decreased start length has been noted in a single third of instances during which it was reported. Round, full face; quick nostril with anteverted nares; upslanting palpebral fissures; lengthy, clean philtrum with skinny, inverted vermilion and wide mouth; thickened helix of usually formed ear; pleasant, happy-natured appearance; gradual coarsening happens postnatally. Markedly brief tubular bones and relatively normal epiphyses; delayed bone age; wide shafts of first and fifth metacarpals and proximal and center phalanges; progressive contractures of a quantity of joints, significantly fingers and wrists; cone-shaped epiphysis, small, irregular capital femoral epiphyses (after 4 years), however other epiphyses, metaphyses, and diaphyses are normal; ovoid vertebral bodies and platyspondyly; J-shaped sella turcica. Shohat M, et al: Geleophysic dysplasia: A storage disorder affecting the pores and skin, bone, liver, heart and trachea, J Pediatr 117:227, 1990. Note brief palpebral fissures, broad nasal bridge, long higher lip, flat philtrum, and thin vermilion border. The boy has small hands and feet, limitation of joint motion, and a "tiptoe" gait. Approximately 40 instances have been reported by 1964 when McKusick and colleagues added fifty two circumstances from an inbred Amish inhabitants. Knee valgus deformities with despair of the lateral tibial plateau and dislocation of the patella need careful orthopedic follow-up. These two genes account for approximately 50% of cases of this dysfunction and each end in an similar phenotype. Ellis�van Creveld syndrome is certainly one of a gaggle of skeletal dysplasias belonging to the ciliopathy group. Disproportionate, irregularly brief extremities; polydactyly of fingers, occasionally of toes; short, broad center phalanges and hypoplastic distal phalanges; malformed carpals, fusion of capitate and hamate, and extra carpal bones; slender thorax with short, poorly developed ribs; hypoplasia of higher lateral tibia, with knock-knee; pelvic dysplasia with low iliac wings and spur-like, downward projections at the medial and lateral elements of the acetabula. Short higher lip sure by frenula to alveolar ridge; defects in alveolar ridge with accessory frenula. Approximately 60% of sufferers have a cardiac defect, most commonly an atrial septal defect; often with a single atrium. Baujat G, Le Merrer M: Ellis-van Creveld syndrome, Orophanet J Rare Dis 2:27, 2007. Verbeek S, et al: Growth charts for kids with Ellis-van Creveld syndrome, Eur J Pediatr one hundred seventy:207, 2011. Note disproportionately short legs and arms, hypoplasia of the alveolar ridge with accessory frenula, polydactyly, hypoplastic fingernails, and, on radiograph, the hypoplastic tibia and the low iliac wings with spur-like, downward projections of the medial and lateral features of the acetabula. Unfortunately, the talipes equinovarus and the scoliosis that develop have been quite resistant to corrective orthopedic measures, and the useful drawback is aggravated by the limitation in joint motility. When present, the unusual defect of hypertrophied auricular cartilage might eventually give way to ossification. Talipes equinovarus plus limitation of flexion at proximal phalangeal joints and of extension at elbow, with or with out dislocation of hip or knee with weight-bearing; short and thick tubular bones with improvement of broad metaphyses and flattened irregular epiphyses which would possibly be late in mineralizing; carpal bones could also be accelerated in ossification in contrast with the rest of the hand; first metacarpal unduly small; abduction of thumbs (hitchhiker thumbs) and nice toes; variable symphalangism of proximal interphalangeal joints; variable webbing at joints. Scoliosis; cervical backbone abnormalities, together with anterior hypoplasia of vertebrae C3 to C5, kyphosis, subluxation, spina bifida occulta, and hyperplastic and dysmorphic odontoid course of; interpedicular narrowing from L1 to L5; accent ossification facilities of manubrium sterni. Soft cystic lots in auricle become hypertrophic cartilage in early infancy in 84% of patients. The mortality rate associated to respiratory obstruction, including laryngeal stenosis, may be as high as 25% in early infancy.
Plendil 5 mg cheap with amexForegut outpouchings and evaginations will now begin to type various glands and the lung and liver primordia blood pressure medication upset stomach plendil 2.5 mg buy discount online. The somites prehypertension medication buy plendil 10 mg on-line, which will differentiate into myotomes (musculature), dermatomes (subcutaneous tissue), and sclerotomes (vertebrae), are evident on into the tail bud. To the proper of that is the creating eye with the optic cup (arrow) and the early invagination of the future lens from surface ectoderm. The free mesenchyme of the limb bud, interacting with the thickened ectodermal cells at its tip, carries all of the potential for the complete growth of the limb. The mesonephric ducts, fashioned within the mesonephric ridges, talk to the cloaca, which is beginning to turn out to be septated, and the yolk sac is regressing. The retina is now pigmented, nonetheless incompletely closed at its inferomedial margin. The auricular hillocks are forming the early auricle (arrow) from the adjoining borders of the mandibular and hyoid swellings. The hand plate (H) has fashioned with condensation of mesenchyme into the five finger rays. The ureteral bud from the mesonephric duct has induced a kidney from the mesonephric ridge, which can additionally be forming gonads and adrenal glands. Cloacal septation is nearly complete; the infraumbilical mesenchyme has filled in all of the cloacal membrane besides the urogenital space; and the genital tubercles are fused, whereas the labioscrotal swellings are unfused. The gut is elongating, and a loop of it may be seen projecting out into the body stalk. In situ embryo (left) with the amnion eliminated (right) to present the outstanding extent of early mind development with formation of the cerebral hemispheres, large coronary heart, nonetheless "paddle-like" limbs, and the regressing tail. The nostril (N) is relatively flat, and the exterior ear (E) is gradually shifting in relative position as it continues to develop and develop. A neck space is now evident, the anterior body wall has formed, and the thorax and stomach are separated by the septum transversum (diaphragm). The phallus and lateral labioscrotal folds are the identical for each sexes at this age. Muscles are developed and functional, regular morphogenesis of joints relies on movement, and first ossification is happening in the centers of growing bones. In the male, the testicle has produced androgen and masculinized the exterior genitalia, with enlargement of the genital tubercle, fusion of the labioscrotal folds right into a scrotum, and closure of the labia minora folds to kind a penile urethra, these structures being unchanged within the female. The pores and skin is growing in thickness, and its accent constructions are differentiating. The form of the palmar floor of the hand and foot, particularly the character of the distinguished apical and different pads, will influence the patterning of parallel dermal ridges that type transversely to the relative lines of progress stress on the palms and soles between 16 and 19 weeks. Serious errors in early morphogenesis seldom permit for survival; hence, only a few malformation problems are seen that may be mentioned to have occurred earlier than 23 days. The cyclopia-cebocephaly sort of defect seems to be the consequence of a defect in the prechordal Table 2-1 Tissues Central nervous system mesoderm, and presumably developed before 23 days. Aside from this instance, the overwhelming majority of serious malformations represent errors that happen after three weeks of improvement. Nilsson L, Ingelman-Sundberg A, Wirsen C: A Child Is Born, New York, 1986, Dell Books. However, the ability of an individual to reach his or her genetic potential with respect to structure, development, or cognitive improvement is affected by environmental elements in each prenatal and postnatal life. Review of the etiologies of these structural abnormalities and syndromes for which an etiology is understood signifies that the majority of malformations and syndromes appear to be genetically determined. The function of this chapter is to outline the most prevalent mechanisms via which genetic abnormalities impression morphogenesis, to talk about the strategies which may be presently obtainable for genetic testing, to counsel genetic counseling for every of these abnormalities, and to talk about approaches to prevention. The haploid human genome incorporates just over 20,000 protein-coding genes, which are far fewer than had been anticipated before sequencing. The great majority of these genes are distributed in the 46 chromosomes which would possibly be found within the nucleus of the cell. A few genes reside within the cytoplasm contained in the mitochondria, the energy-producing equipment of the cell. The frequency with which each of those genetic mechanisms contributes to malformation and illness is dependent upon the time in growth at which inquiry is made. For instance, roughly half of all first-trimester miscarriages are a consequence of chromosomal abnormalities, whereas solely 6 of one thousand live-born infants are equally affected. Each of these issues is taken into account separately as it pertains to malformation, especially a number of defect syndromes. Normal improvement is dependent not solely on the gene content of those chromosomes but on the gene stability as well. An altered variety of chromosomes most commonly arises because of fault in chromosome distribution at cell division. Abnormal segregation in meiosis or mitosis will lead to an incorrect variety of chromosomes (aneuploidy) in daughter cells. In addition, a bit of a chromosome may be deleted, duplicated, inverted, or exchanged between two chromosomes. It would clearly be troublesome to rely these chromosomes or to distinguish their individual structure from such preparations. The dots throughout the chromosomes symbolize "normal" genes, the bar represents a dominant mutant gene, the hash-bar represents a recessive mutant gene, and the triangles denote main and minor genes that confer susceptibility to a given process. B, Metaphase cell with chromosomes hooked up to the spindle fibers and beginning to separate. C, Anaphase cell with similar chromosomal complements having been "pulled aside" toward the event of two daughter cells. Various strategies, similar to trypsin therapy and Giemsa staining, can be used to permit for the identification of individual chromosomes. The growth of synchronized tradition techniques that permit evaluation of chromosomes in prophase and prometaphase have tremendously enhanced the power to detect delicate abnormalities and have expanded our understanding of the impression of chromosomal rearrangement on morphogenesis. Banding strategies utilized on metaphase or prometaphase preparations enable the popularity of each of the person chromosomes, aneuploidies, and loss or gain of chromosome fragments larger than 5 Mb in normal resolution and 3 Mb in high-resolution karyotypes. When viewed with a wavelength of sunshine that excites the fluorescent dye, a coloured signal is generated, permitting localization of the probe. Giemsa-stained chromosomes organized right into a karyotype by letter grouping and number designation on the idea of size of the chromosome, place of the centromere, and banding patterns. Giemsa-stained chromosome quantity 2 harvested at totally different points in the cell cycle. The two are blended in equal amounts and "painted" on regular human chromosome preparations. Deviations from the anticipated 1: 1 ratio of red to green shall be detected as a change within the shade signal in that region documenting achieve or lack of copy number. If the printed sequences overlap, in a so-called tiling-path array, coverage of the entire genome could additionally be achieved.
Generic plendil 10 mg with mastercardIn general hypertension quiz 2.5 mg plendil generic otc, all daughters of provider moms might be clinically normal prehypertension define plendil 2.5 mg buy cheap line, although 50% will carry the altered gene and have a danger for vertical transmission. The identification of earlier affected males associated by way of potential provider females is a strong argument for X-linked inheritance. X-inactivation research in the moms of the affected male may reflect markedly skewed X-inactivation (>95%), which helps this mode of inheritance. Mothers of daughters with X-linked dominant circumstances should be examined intently for proof of medical impact. If the mother is regular, recent gene mutation in the offspring is likely and the risk for recurrence is negligible. Females, then again, have a risk that approaches 100 percent, as a result of the human egg is the source of the entire mitochondria for the offspring. Most affected women have each regular and irregular mitochondria; thus, any given egg will have Genetics, Genetic Counseling, and Prevention 887 each sorts in numerous proportions. Random distribution of mitochondria in dividing cells within the early embryo creates completely different proportions of abnormal to normal mitochondria in several tissues. A medical phenotype happens only when a threshold of irregular to normal mitochondria is exceeded in a crucial tissue. Thus, all offspring of affected girls could additionally be assumed to have inherited some abnormal mitochondria; however, not all will manifest disease. The multifactorial/threshold model makes several predictions that in large measure are in accord with the clinical and epidemiologic observations relating to given malformations: 1. In addition, many common malformations have different start frequencies in numerous populations. The danger for first-degree family members (parents, siblings, and offspring) approximates the sq. root of the inhabitants danger. For nearly all of defects, the risk is 2% to 5%, which is 20 to forty instances the frequency of the issue within the general inhabitants. The figures in Table 3-1 are derived from direct observations in clinical populations and correlate properly with the numbers predicted by the mannequin. Second-degree relations (uncles, aunts, halfsiblings) have a sharply lower danger than first-degree family members. The mannequin concerned the concept of genetic liability or susceptibility to a given attribute, governed by many different genes, and a threshold, decided by both genetic and environmental elements. The model transformed the traditional distribution of a morphogenetic process within a population into an "all-or-none" expression of a structural defect. Normal Parents of One Affected Child 4%�5%* 2%�6% 3%�4% 3% 3%�5% 2%�8% 3%�4% 3%�5% 10%�15% Future Males Future Females 4% 2. The higher the quantity is of affected members of the family, the larger the danger is for recurrence. This sample of recurrence of multifactorial traits is in distinction to each dominant and recessive inheritance by which the chance for future offspring remains unchanged regardless of recurrences. This idea relates to the fact that inbreeding will increase the number of "susceptibility genes," thus making a developmental drawback more probably. This presumes that the severity of the malformation reflects a higher adverse genetic influence, thereby rising the risk from the same parentage. Certainly with cleft lip and palate, data help the hypothesis, as a result of the danger for recurrence in subsequent kids when an offspring has a extreme bilateral cleft lip and palate is 5. The risk for recurrence shall be elevated for family members of the least affected gender, if gender variations are famous. Some of the gender variations in malformation prevalence could also be explained as the direct effects of structural genital variations, such as hypospadias in the male. Similarly, the marked male predominance of the urethral obstruction sequence may be defined by the reality that the most typical website of urethral obstruction is the prostatic urethra. The humoral impact of testosterone, which makes connective tissue tougher within the male, could clarify the preponderance of dislocation of the hip, related to connective tissue laxity, in the female. Also, testosterone, which is produced by the male in the course of the first few postnatal months, may improve the likelihood of muscle hypertrophy and thereby increase the tendency to develop hypertrophic pyloric stenosis. The gender variations related to the incidence of other structural defects would seem to imply that genes on the X or Y chromosome could increase the likelihood of particular anomalies creating during morphogenesis. Thus, excluding anomalies associated to joint laxity, most late uterine constraint-induced deformations are more frequent in the male, who is often rising faster within the last trimester of gestation than the feminine. It is hypothesized that if it takes more genetic elements to give rise to an anomaly within the female, then the affected female should cross on more of these genetic elements to her offspring, who would have a better frequency of the anomaly than would offspring of affected males. Observational research in pyloric stenosis documenting a 24% danger of transmission from affected moms compared to 6% from affected fathers bear this out. The frequency of concordance and discordance in monozygotic and dizygotic twins has been used to argue each for environmental and for singlegene causation of common malformations. For most of these defects, the incidence of concordance in dizygotic twins is similar to that of siblings born of separate pregnancies, Proportion of Males vs Females Pyloric stenosis Clubfoot Cleft lip � palate Cleft palate alone Meningomyelocele Anencephaly Cong. Genetics, Genetic Counseling, and Prevention 889 arguing towards each a single-gene etiology and a serious environmental influence. Over the previous 15 years, numerous investigators have reanalyzed previously published data units for particular "multifactorial" malformations with respect to quite a lot of different hypotheses. For cleft lip with or with out cleft palate, it now seems probably that susceptibility is set by a small variety of genes (two to eight) performing in a multiplicative style and interacting with environmental factors. This is most likely going true with respect to genes that outline susceptibility to different widespread "multifactorial" malformations. Despite the accumulating proof that suggests that the multifactorial mannequin is probably not strictly biologically true, the empiric knowledge obtained from the observational studies used to construct the model remain the premise for genetic counseling of households. Molecular testing for susceptibility genes might be attainable when entire genome sequencing turns into clinically out there. That environmental influences play a task in the willpower of widespread malformations is borne out by many studies similar to these of anencephaly and meningomyelocele that doc that social class is a variable that impacts delivery frequency. Birth-order influences have also been noted, with congenital dislocation of the hip and pyloric stenosis being extra likely to occur in firstborn youngsters. One apparent environmental factor is fetal in utero constraint resulting in deformation. Environmental elements such as this most likely explain the larger frequency of dislocation of the hip in addition to most other deformations within the firstborn. Studies in experimental animals have dramatically illustrated the profound influence that genetic background could have on the likelihood of a given environmental teratogen inflicting malformation. For example, Fraser could regularly produce cleft palate in mouse embryos of the A/Jax pressure by giving the moms a excessive dose of cortisone throughout early gestation, whereas the same remedy in a unique strain led to solely 17% affected offspring. In humans, genetic susceptibility to hydantoininduced teratogenesis appears to correlate with the genetically decided activity ranges of epoxide hydrolase, one of many enzymes essential for the metabolism of hydantoin. Expression of the phenotype requires both genetic susceptibility and drug exposure. The search for environmental factors that enable for expression of a single malformation is ongoing.
Blighia sapida (Ackee). Plendil. - Are there safety concerns?
- How does Ackee work?
- Dosing considerations for Ackee.
- Colds, fever, water retention, and epilepsy.
- What is Ackee?
Source: http://www.rxlist.com/script/main/art.asp?articlekey=96793
5 mg plendil buy with amexCytotoxics: possibly elevated unwanted side effects of cyclophosphamide; cut back dose of ruxolitinib heart attack chords buy plendil 5 mg cheap. Diuretics: elevated eplerenone ranges � keep away from concomitant use; focus of fluconazole elevated by hydrochlorothiazide blood pressure pills kidneys buy 5 mg plendil overnight delivery. Cytotoxics: elevated pulmonary toxicity with pentostatin (unacceptably high incidence of fatalities); increases intracellular concentration of cytarabine. Clearance of fludarabine from the plasma is triphasic; elimination is usually via renal excretion: 40�60% of an intravenous dose is excreted in the urine. Administer up to achievement of scientific response (usually 6 cycles) then discontinue. The pharmacokinetics and pharmacodynamics of fludarabine phosphate in sufferers with renal impairment: a prospective dose adjustment research. In human volunteers, excretion by way of urine was about 80%, and it was concluded that about 20% was excreted by a unique route. It is likely that, as for the metabolism of different steroids, excretion into the bile is balanced by reabsorption in the gut and a few half is excreted with the faeces. Antibacterials: metabolism accelerated by rifamycins; metabolism presumably inhibited by erythromycin; probably scale back isoniazid focus. The carboxylic acid metabolite is the primary metabolite in plasma (free form) and urine (free kind and its glucuronide). This major metabolite showed no benzodiazepine agonist or antagonist activity in pharmacological tests. Practically no unchanged flumazenil is excreted in the urine, suggesting complete metabolic degradation of the drug. Elimination of radiolabelled drug is actually full inside 72 hours, with 90�95% of the radioactivity showing in urine and 5�10% in the faeces. The half-life of flumazenil is shorter than these of diazepam and midazolam � sufferers ought to be intently monitored to keep away from the chance of them becoming re-sedated. It is distributed throughout body tissues and fluids, and disappears from the plasma inside about three hours. About 15% of an intravenous dose is excreted unchanged in the urine within 6 hours. The elimination half-life of fluoxetine is 4 to 6 days and for norfluoxetine four to sixteen days. Anti-epileptics: antagonism (lowered convulsive threshold); concentration of carbamazepine and phenytoin increased. Antipsychotics: focus of haloperidol, clozapine and risperidone elevated; probably inhibits aripiprazole metabolism � scale back aripiprazole dose; increased risk of ventricular arrhythmias with droperidol and pimozide � avoid. Hormone antagonists: metabolism of tamoxifen to lively metabolite probably reduced � avoid. Antipsychotics: keep away from concomitant use of clozapine with depot preparations in case of neutropenia. Paths of metabolism of flupentixol include sulfoxidation, side-chain N-dealkylation, and glucuronic acid conjugation. The price of urinary excretion of flurbiprofen and its two main metabolites ([2-(2-fluoro4-hydroxy-4-biphenylyl) propionic acid] and [2-(2-fluoro-3-hydroxy-4-methoxy-4biphenylyl) propionic acid]) in both free and conjugated states is analogous for both the oral and rectal routes of administration. Antivirals: focus presumably elevated by ritonavir; increased threat of haematological toxicity with zidovudine. The main parts circulating in the blood are fluvastatin and the pharmacologically inactive N-desisopropylpropionic acid metabolite. About 93% is excreted in the faeces, mainly as metabolites, with solely about 6% being excreted within the urine. Anti-epileptics: focus of both or each medication could additionally be elevated with phenytoin. Ciclosporin: concomitant therapy with ciclosporin might result in threat of muscle toxicity. Lipid-lowering medication: increased risk of myopathy with gemfibrozil, fibrates and nicotinic acid � keep away from concomitant use with gemfibrozil. The Committee on Safety of Medicines has suggested that rhabdomyolysis related to lipid-lowering medication, such because the fibrates and statins, seems to be rare (approx. Excretion is principally in the urine; about 2% of a dose is excreted as unchanged drug. Anti-epileptics: antagonise anticonvulsant threshold; focus of carbamazepine and phenytoin elevated. Antipsychotics: focus of asenapine, haloperidol, clozapine and olanzapine elevated; elevated threat of ventricular arrhythmias with droperidol and possibly pimozide � keep away from. Theophylline: elevated theophylline concentrations � keep away from concomitant use; if not attainable, halve theophylline dose and monitor ranges. It is converted to the metabolically energetic kind 5-methyltetrahydrofolate in the plasma and liver. Folate metabolites are eradicated within the urine and folate in excess of body requirements is excreted unchanged within the urine. Manufacturer advises to keep away from in extreme renal impairment as a outcome of increased risk of bleeding. Anticoagulation with fondaparinux for hemodiafiltration in patients with heparin-induced thrombocytopenia: dose-finding research and security evaluation. Turbohaler: 6�24 mcg 1�2 instances every day as a single dose, up to seventy two mcg daily could also be needed. Minor pathways involve sulphate conjugation of formoterol and deformylation adopted by sulphate conjugation. After a single oral dose of 3H-formoterol, 59�62% of the dose was recovered in the urine and 32�34% in the faeces. The main route of metabolism of amprenavir is through the cytochrome P450 3A4 enzyme. The main route of elimination of amprenavir is through hepatic metabolism with less than 1% excreted unchanged within the urine and no detectable amprenavir in faeces. Metabolites account for approximately 14% of the administered amprenavir dose in the urine, and approximately 75% within the faeces. Potentially hazardous interactions with other medicine Anti-arrhythmics: presumably elevated concentration of amiodarone, flecainide, lidocaine and propafenone (increased danger of ventricular arrhythmias) � avoid concomitant use. Antibacterials: will increase focus of rifabutin � reduce rifabutin dose; focus considerably reduced by rifampicin � avoid concomitant use; keep away from concomitant use with telithromycin in severe renal and hepatic impairment. Antimalarials: use artemether/ lumefantrine with caution; possibly will increase quinine focus. Antipsychotics: possibly inhibits aripiprazole metabolism � cut back aripiprazole dose; possibly increases quetiapine focus � keep away from; possibly increases pimozide concentration (increased threat of ventricular arrhythmias) � keep away from concomitant use.
Buy 5 mg plendil otcThe decrease portion forms compartments for the flexor digitorum longus and flexor hallucis longus muscle tissue pulse pressure for athletes order 10 mg plendil free shipping. The tibial nerve and posterior tibial artery and vein lie between the two membranous elements arterial dissection generic plendil 10 mg on line. Usually cruciate band that supports the extensor tendons, extending from each malleoli to the foot margins of the opposite aspect, primarily to the calcaneus. Upper band that holds peroneal tendons in place; it extends from the lateral malleolus to the calcaneus. Synovial bursa between the pterygoid hamulus and the tendon of the 18 tensor veli palatini muscle. Synovial bursa between the pores and skin and the laryngeal prominence of the thyroid cartilage. Synovial bursa between the body of the hyoid bone and the median thyrohyoid ligament. Synovial bursa between the higher finish of the sternohyoid muscle and the thyrohyoid membrane. Synovial bursa between the biceps tendon and the anterior part of the radial tuberosity. Common tendon sheath forming the first tendon compartment on the dorsum of the hand. Common tendon sheath forming the second tendon compartment on the dorsum of the hand. Synovial bursa at the attachment between the tendon and base of the 3rd metacarpal. Synovial bursa between the trapezius muscle (ascending part) and the backbone of the scapula. Synovial bursa between the acromion, coracoacromial ligament and supraspinous tendon. Synovial bursa between the deltoid muscle and the larger tubercle of the humerus. Synovial bursa between the tendons of the subscapularis and coracobrachialis muscles under the apex of the coracoid process. Synovial bursa between the tendon of the infraspinatus and the capsule of the shoulder joint. Synovial bursa between the tendon of the subscapularis and the capsule of the shoulder joint. Individual tendon sheath for the flexor carpi radialis on the insertion of the tendon to the 16 base of the 2nd metacarpal bone. Synovial bursa between the origins of the biceps femoris and semimembranosus muscle tissue. Synovial bursa between the investing fascia of the knee and the tendon of the quadratus femoris muscle. Synovial bursa immediately on the knee joint beneath the tendon of the quadratus femoris. Synovial bursae between the sartorius tendon and the tendons of the gracilis and semitendinosus located beneath it. Synovial bursa on the tendon of the gluteus maximus between the skin and higher trochanter. Synovial bursa between the tendon of the gluteus maximus and the higher trochanter. This designation comprises two synovial bursae, an anterior one between the tendon of insertion of the gluteus medius and the higher trochanter and a posterior one between this tendon and the piriformis muscle. Synovial bursa between the tendon of insertion of the gluteus minimus and the greater trochanter. Synovial bursa between the cartilage-covered surface of the lesser sciatic notch and the tendon of the obturator internus. Synovial bursa between the ischial tuberosity and the inferior surface of the gluteus maximus. Synovial bursa on the tibial collateral ligament below the tendons of the semitendinosus, gracilis and sartorius muscle tissue. Synovial bursa located partially on the fibular collateral ligament under the tendon of insertion of the biceps femoris. Synovial bursa on the lateral femoral condyle beneath the tendon of origin of the popliteal muscle. It at all times communicates with the knee joint cavity, more not often with the tibiofibular joint. Synovial bursa between the lateral condyle of the femur and the lateral gastrocnemius tendon. Synovial bursa between the medial condyle of the femur and the medial gastrocnemius tendon. Synovial bursa between the semimembranosus tendon and the upper margin of the tibia. It extends as a lot as the proximal finish of the only, where it crosses under the tendon of the flexor digitorum longus. Synovial bursa between the tibialis anterior tendon and the medial cuneiform bone. Mucosal fold overlying the sublingual gland and lengthening posterolaterally from the sublingual papilla. Small mucosal elevation at the opening of the parotid duct lateral to the second higher molar tooth. Small mucosal elevation over the incisive foramen on the anterior end of the palatine raphe. Salivary glands similar to the buccal glands located beneath the mucosal at the stage of the molar teeth. Numerous mucous, serous and combined glands primarily within the lateral and posterior areas of the tongue. True oral cavity enclosed anteriorly and laterally by the teeth and increasing so far as the isthmus of fauces (oropharyngeal isthmus). Median mucosal ridge on the junction of the proper and left bony palatal processes. Mucous membrane of oral cavity consisting of stratified, nonkeratinized squamous epithelium throughout and underlying mixed glands. Mixed glands close to the apex of the tongue providing several drainage ducts on the undersurface of the tongue. About 40 small ducts that drain the sublingual gland and open alongside the sublingual fold and the sublingual caruncle. It loops around the posterior margin of the mylohyoid accompanied by glandular tissue and opens on the sublingual caruncle. Portion of the parotid gland located on the masseter muscle near the parotid excretory duct. It passes around the anterior margin of the masseter and opens close to the second upper molar tooth. Distinct eminence on the aspect of the crown, particularly in canine and incisor enamel.

Buy plendil 2.5 mg visaSandrow and colleagues described a similarly affected father and daughter who had hypertension and pregnancy purchase plendil 5 mg on line, in addition heart attack indigestion plendil 10 mg lowest price, anomalies of the ala nasi and columella. An affected 54-year-old male with mild intellectual incapacity who appeared older than his age was described as cheerful and apparently wholesome. Deep groove operating the size of a short columella, flat nasal bridge, bulbous nasal tip, unfused nares, hypoplastic alar and columellar cartilage. Complete polysyndactyly; cupappearing, rosebud, or mitten arms; phalanges of differing dimensions and shapes; disorganized interphalangeal joints; abnormal carpal bones. Polydactyly with variable syndactyly, mirror image feet, talipes equinovarus, irregular tarsal bones, absent/hypoplastic tibia. Large mandibular condyles; duplication of ulna; malformed scaphoid and lunate bones; absence of the trapezia, triquetrum, and pisiform bones; synostosis/malformation of tarsals; synostosis of talus, calcaneus, cuboid, and navicular bones; supernumerary metacarpals and metatarsals; uneven shortening of metacarpals; bony syndactyly of phalanges; radioulnar synostosis; absent/hypoplastic patella. Mari�o-Enr�quez A, et al: Laurin-Sandrow syndrome: Review and redefinition, Am J Med Genet A 146A:2557, 2008. Note frontal bossing, hypertelorism, broad nasal bridge, flat midface and nostril with a deep longitudinal groove on the tip of the nostril and the columella, downturned corners of mouth. Note bilateral polysyndactyly with bilateral postaxial appendices and a thumb-appearing preaxial finger. Seven metacarpals and finger anomalies are noted on radiographs of the upper limbs. B, Radiographic look displaying four lateral toes with three identically shaped phalanges, and different phalanges displaying variable styles and sizes. On the radiograph of the lower limbs, observe the hypoplastic tibiae, that are shorter than the fibula. With the exception of kind V, all have related oral, facial, and digital abnormalities. Significant overlap exists between the thirteen sorts, making it troublesome to present applicable counseling relative to prognosis. Type V (Thurston syndrome), an autosomal recessive situation, includes midline cleft lip, duplicated frenulum, and postaxial polydactyly of arms and ft. Occasional options embody development hormone deficiency, hypogonadotrophic hypogonadism, and a hypothalamic hamartoma. The latter findings are features of Pallister-Hall syndrome and raise the possibility that these two issues are the same. Multiple and/or hyperplastic frenuli between the buccal mucous membrane and alveolar ridge, median cleft lip, lobated/bifid tongue with nodules, cleft of alveolar ridge (at area of lateral incisors, which may be missing), cleft palate, dental caries, and anomalous anterior tooth. Hypoplasia of alar cartilages, lateral placement of inner canthi; milia of ears and upper face in infancy. Asymmetric shortening of digits with clinodactyly, syndactyly, or brachydactyly of arms and preaxial polydactyly of toes. Increased naso-sella-basion angle at base of skull; absence of corpus callosum; intracerebral cyst; porencephaly; hydrocephalus; vermis hypoplasia; focal polymicrogyria; cortical, periventricular, subarachnoid heterotopia; and Dandy-Walker malformation. Management is directed toward plastic surgical correction of oral clefts and dental care, together with dentures when indicated. Psychometric analysis is merited as a outcome of about one half of the reported patients have intellectual incapacity. Polycystic renal illness is progressive, with onset of hypertension and renal insufficiency after 18 years of age. Fibrocystic Oral-Facial-Digital Syndrome 353 distinguish this condition embody congenital hydronephrosis, coarse hair, facial asymmetry, facial weakness, and preauricular tags. Features that distinguish this condition are retinal coloboma and hallucal duplication. References Papillon-L�age E, Psaume J: Une malformation h�r�ditaire de la muqueuse buccale: Brides et freins anormaux, Rev Stomatol (Paris) fifty five:209, 1954. Majewski F, et al: Das oro-facio-digitale Syndrom: Symptome und Prognose, Z Kinderheilkd 112:89, 1972. Donnai D, et al: Familial orofaciodigital syndrome type I presenting as grownup polycystic kidney illness, J Med Genet 24:eighty four, 1987. Gurrieri F, et al: Oral-facial-digital syndromes: Review and diagnostic pointers, Am J Med Genet 143:3314, 2007. A�C, Note the milia of the ears and higher face in infancy, the median cleft lip, and the hypoplastic ala nasi. F and G, Note the uneven shortening of digits with syndactyly and clinodactyly. Low nasal bridge with lateral displacement of inside canthi; broad nasal tip, sometimes slightly bifid; midline partial cleft of lip; hypertrophy of traditional frenula; midline cleft of tongue, nodules on tongue; flare to alveolar ridge; hypoplasia of zygomatic arch, maxilla, and body of mandible. Partial reduplication of hallux and first metatarsal, cuneiform, and cuboid bones; comparatively short palms with clinodactyly of fifth finger; bilateral postaxial polydactyly of arms; bilateral preaxial polysyndactyly of ft (occasionally only unilateral); metaphyseal flaring and irregularity. A�C, Note the midline cleft of the upper lip, lateral displacement of the medial canthi, broad nasal tip, and tongue nodules. D�F, Note the postaxial polydactyly of hands and ft and preaxial polydactyly of toes. The sample of malformation expanded rapidly to embody other defects of the third and fourth branchial arches in addition to dysmorphic facial options. In 1978, Shprintzen and colleagues reported a gaggle of kids with cleft palate or velopharyngeal incompetence, cardiac defects, and a prominent nostril (velo-cardiofacial syndrome). It was subsequently decided that individuals with velo-cardio-facial syndrome and nearly all of those with the situation described by DiGeorge have a deletion of chromosome 22q11. It is now known that the two disorders represent completely different manifestations of the same genetic defect. Speech is almost at all times hypernasal, with the pharyngeal musculature being hypotonic. Personality might tend toward perseverative conduct, with concrete considering secondary to mental impairment or studying issues. Approximately 10% of affected people have developed psychiatric problems, primarily chronic schizophrenia and paranoid delusions, with onset various between 10 and 21 years of age. Obstructive sleep apnea has been famous following pharyngeal surgery to enhance speech in a quantity of sufferers. Cleft of the secondary palate, either overt or submucous; velopharyngeal incompetence; small or absent adenoids; outstanding nostril with squared nasal root and narrow alar base; narrow palpebral fissures; ample scalp hair; deficient malar area; vertical maxillary excess with long face; retruded mandible with chin deficiency; microcephaly (40%�50%). Defects current in 85%, the commonest being ventricular septal defect (62%); proper aortic arch (52%); tetralogy of Fallot (21%); aberrant left subclavian artery. Because of the marked variability of expression, each dad and mom of an affected baby should be tested to decide if they carry the deletion. Choanal atresia and options of the oculoauriculo-vertebral spectrum have also been seen. Ueb2l3 causes severe development retardation in mice and will account for progress retardation in these patients. Distal duplications show low penetrance and marked variable expression for developmental delay. Most people (70%) have inherited the duplication, most commonly from a normal or near-normal father or mother, whereas deletions occur de novo in 90% of circumstances.
Proven 10 mg plendilGersh M hypertension signs cheap plendil 10 mg fast delivery, et al: Evidence for a distinct area inflicting a cat-like cry in patients with 5p deletions arteria femural plendil 2.5 mg purchase amex, Am J Med Genet 56:1404, 1995. However, it is necessary to realize that del 9p is heterogeneous and is associated with variable deletion sizes. The candidate area for intercourse reversal, an occasional characteristic of deletion 9p syndrome, has been narrowed all the method down to the 9p24. Craniosynostosis involving the metopic suture leading to trigonocephaly; flat occiput; short, upslanting palpebral fissures; epicanthal folds, distinguished eyes secondary to hypoplastic supraorbital ridges; highly arched eyebrows; midfacial hypoplasia with a brief nostril, depressed nasal bridge, anteverted nares, and long philtrum; small mouth, micrognathia; posteriorly rotated, poorly formed ears with hypoplastic, adherent ear lobes; short broad neck with low hairline. Long middle phalanges of the fingers with extra flexion creases; short distal phalanges with brief nails; extra in whorl patterns on fingertips; foot positioning defects; simian crease. Ventricular septal defects, patent ductus arteriosus, and/or pulmonic stenosis in a single third to one half of patients. Scoliosis, broadly spaced nipples, diastasis recti, inguinal and/or umbilical hernia, micropenis and/or cryptorchidism in males; hypoplastic labia majora in females. Onesimo R, et al: Chromosome 9 deletion syndrome and sex reversal: novel findings and redefinition of the critically deleted areas, Am J Med Genet 158A:2266, 2012. Chilosi A, et al: Del (9p) syndrome: Proposed behavior phenotype, Am J Med Genet 100:138, 2001. A and B, Note the prominent brow with metopic ridge, trigonocephaly, frontal hair upsweep, quick nostril with anteverted nares, and low-set ears. Although intellectual incapacity occurs in just about all of those patients, the severity of the scientific phenotype correlates with the extent of the triplicated material. Partial trisomy 9pterp21 is associated with delicate craniofacial options and uncommon skeletal or visceral defects. Partial trisomy 9pterp11 is related to the everyday craniofacial features, whereas partial trisomy 9pterq11-13 is related not solely with the typical craniofacial options but also with skeletal and cardiac defects. Partial trisomy 9pterq22-32 is associated with the typical craniofacial options, intrauterine progress deficiency, cleft lip/palate, micrognathia, cardiac anomalies, and congenital hip dislocation. If the trisomic segment is larger than that (9pter9q31 or 32), the clinical findings not match into the trisomy 9p syndrome however somewhat resemble trisomy 9 mosaic syndrome. Growth deficiency, primarily of postnatal onset; delayed puberty such that some patients proceed to grow up to the middle of their third decade. Microcephaly, hypertelorism, downslanting palpebral fissures, deep-set eyes, distinguished nostril, downturned corners of the mouth, cup-shaped ears. Short fingers and toes with small nails and short terminal phalanges; fifth finger clinodactyly with single flexion crease; single palmar crease. Kyphoscoliosis, normally developing during the second decade; hypoplasia of periscapular muscle tissue with deep acromial dimples; defective ossification of the pubic bone, broad ischial tuberosity; pseudoepiphysis of metacarpals, metatarsals, and center phalanges of fifth fingers; delayed closure of cranial sutures and fontanels. Schinzel A: Trisomy 9p, a chromosome aberration with distinct radiologic findings, Radiology one hundred thirty:a hundred twenty five, 1979. Pseudoepiphyses on metacarpals and metatarsals 2 to 5; notches on metacarpal 1, metatarsal 1, and proximal and center phalanges of fingers; hypoplasia of the center phalanx of fifth finger, terminal phalanges of fingers, and center and terminal phalanges of toes; thick epiphyses, particularly of terminal phalanges of massive toe, thumb, and little finger; and clinodactyly of fifth finger. A�D, Note the ocular hypertelorism, prominent nostril, downturned corners of mouth, cup-shaped ear, quick fingers, and 3-4 syndactyly. Surviving children confirmed marked mental deficiency and usually are bedridden without the ability to talk. Individuals with dup10q25qter lack major malformations, and the prognosis is more favorable. Microcephaly; flat face with excessive forehead and excessive, arched eyebrows; ptosis; short, downslanting palpebral fissures; microphthalmia; broad and depressed nasal bridge, anteverted nares, bow-shaped mouth with outstanding upper lip; cleft palate; malformed posteriorly rotated ears. Camptodactyly, proximally positioned thumbs, syndactyly between second and third toes, foot place anomalies, hypoplastic dermal ridge patterns. Heart and renal malformations, every of which occurs in roughly one half of affected patients; kyphoscoliosis; pectus excavatum; 11 pairs of ribs; congenital hip dislocation; cryptorchidism. Briscioli V, et al: Trisomy 10qter confirmed by in situ hybridization, J Med Genet 30:601, 1993. Note the flat face with excessive brow; broad nasal bridge; anteverted nares; malformed, posteriorly rotated ears; camptodactyly; and proximally placed thumbs. Familial occurrence ensuing from unbalanced transmission of a balanced insertional translocation has been recorded. An interstitial deletion in 11p must be particularly sought within the cytogenetic investigation of intellectually disabled patients with Wilms tumor and/or aniridia. However, a case could be made for frequent stomach examination and periodic testing into adulthood with frequent screening for hypertension and proteinuria. Moderate to severe mental disability in most patients, attention-deficit/ hyperactivity dysfunction, autism spectrum disorder. Differences within the dimension of the deleted section (especially distal to 11p13) in individual circumstances could account for the noticed variability in concomitant features and within the diploma of progress and psychological deficiency. A and B, Prominent lips and poorly fashioned ears in a feminine with aniridia�Wilms tumor association. However, it appears that the scientific phenotype is due to deletion of subband 11q24. Feeding difficulties are widespread, and persistent constipation happens in almost 50% of those kids. Although all degrees have been reported, roughly one half have been within the average range, and a lot of the remaining are more severely affected. Hypotonia in infancy, frequently progressing toward spasticity, hearing loss, speech impairment. Prominent brow (62%), microcephaly (40%), epicanthal folds (60%), ocular hypertelorism (70%), ptosis (67%), strabismus (75%), depressed nasal bridge (93%), brief nostril with upturned nasal tip (91%) and lengthy philtrum, giant, carp-shaped mouth (78%) with thin upper lip, micrognathia (77. Joint contractures (65%); cardiac defect (60%), primarily ventricular septal defect and left-sided obstructive defect, hypospadias and/ or cryptorchidism (50%); Paris-Trousseau syndrome (defect in platelet development characterized by neonatal thrombocytopenia and persistent platelet dysfunction). Schinzel A, et al: Partial deletion of lengthy arm of chromosome 11[del(11)(q23)]: Jacobsen syndrome, J Med Genet 14:438, 1977. Subsequently, greater than one hundred thirty cases have been recorded, and the deleted chromosome has been thought-about number thirteen. Although the phenotype is variable, it might be divided into three clusters based mostly on the particular deleted phase of 13q. If the q14 region is involved, a significant threat exists for retinoblastoma; intellectual disability; progress deficiency; and main malformations, together with microcephaly and central nervous system defects, distal limb anomalies, eye defects, and gastrointestinal malformations. Patients with essentially the most distal deletions, involving q33-q34, have extreme mental incapacity but usually lack progress deficiency or gross structural malformations although a case with uncontrolled epilepsy has been described. Mental deficiency, microcephaly with tendency toward trigonocephalyand holoprosencephaly-type brain defects. Prominent nasal bridge, hypertelorism, ptosis, epicanthal folds, microphthalmia, colobomata, retinoblastoma (usually bilateral), prominent maxilla, micrognathia, prominent, slanting, low-positioned ears.
5 mg plendil discount with visaFraccaro M: Contributo allo studio delle malattie del mesenchima osteopoietico: I achondrogenesi hypertension glaucoma plendil 5 mg buy with visa, Folia Hered Pathol (Milano) 1:a hundred ninety blood pressure 3 readings generic 5 mg plendil mastercard, 1952. Maroteaux P, Lamy M: Le diagnostic des nanismes chondrodystrophiques chez les nouveau-n�s, Arch Fr Pediatr 25:241, 1968. Borochowitz Z, et al: Achondrogenesis type I-further heterogeneity, J Pediatr 112:23, 1988. Large calvarium with large anterior and posterior fontanels, flat nasal bridge, small anteverted nostrils, micrognathia. Normal cranial ossification; quick ribs with out fractures; brief, broad lengthy bones with disproportionately lengthy fibula and metaphyseal irregularity of distal ulna; variable levels of failure of ossification of lumbar spine, cervical backbone, sacrum, ischial and pubic bones, and calcaneus and talus. A variety of examples of multiple affected youngster in a family born to unaffected mother and father have been reported and are felt to be the results of germline mosaicism. Wainwright H, Beighton P: Visceral manifestations of hypochondrogenesis, Virchows Arch 453:203, 2008. Note the comparatively normal cranial ossification, brief ribs, and variable degrees of failure of ossification of lumbar and cervical spines, sacrum, and ischial and pubic bones. A distinctive fibrosis of the growth-plate cartilage led to the designation fibrochondrogenesis. Widely patent anterior fontanel, coronal and sagittal sutures; protuberant eyes with giant corneae; hypoplastic nose with flat nasal bridge and anteverted nares; lengthy philtrum; small mouth; cleft palate; short neck; low-set, malformed ears. Flattened vertebrae with posterior vertebral hypoplasia and a sagittal midline cleft; short, thin ribs with anterior and posterior cupping; long, skinny clavicles; small chest; small/elevated scapula. Rhizomelic shortening; small arms and toes; camptodactyly; fifth-finger clinodactyly; hypoplastic finger and toenails; quick, dumbbellshaped long bones with broad, irregular metaphyses; prominent metaphyseal spurs adjoining to development plates; short fibulae. Hypoplastic with ovoid ilia, irregular flattened acetabula with medial spikes and slender sacrosciatic notches; broad, hypoplastic ischii. References Lazzaroni-Fossati F, et al: La fibrochondrogenese, Arch Fr Pediatr 35:1096, 1978. Bankier A, et al: Fibrochondrogenesis in male twins at 24 weeks gestation, Am J Med Genet 38:ninety five, 1991. Note the flat, broad nasal bridge, anteverted nares, short limbs, and equinovarus position of the feet. B, the radiograph reveals long, skinny clavicles; quick, thin ribs; flattened acetabula; narrow sacrosciatic notches; metaphyseal widening of the tibia and fibula; and dumbbell-shaped femora. There is shortening of proximal and distal bones of the limbs, radial deviation of the thumb, and a big hole between toes 1 and a pair of. Neonatal demise is uniform and related to pulmonary hypoplasia, tracheobronchomalacia, and a malformed stenotic larynx. Radiographic features include platyspondyly, cervical kyphosis, hypoplasia/dysplasia of vertebrae, quick ribs, glenoid hypoplasia, horizontal acetabulae, bifid or V-shaped humerus, rounded distal femora, bowing of radius and tibia, and hypoplasia/dysplasia of tubular bones of palms and ft. Inactivation of the gene product, a sulfate-chloride exchanger of the cell membrane, results in intracellular sulfate depletion and to synthesis of undersulfated proteoglycans in susceptible cells. This early lethal short-limbed dwarfing condition was set forth by Maroteaux and colleagues and Sillence and colleagues. Atelosteogenesis derives from the Greek word for "incomplete" and relates to the marked lack of full ossification of sure bones. Humeri are absent, segment-shaped, or distally tapered; absent fibula; quick distally pointed femora; bowed tibiae; abnormally segmented and fused cervical vertebrae; thoracic platyspondyly with a quantity of coronal clefts throughout; eleven pairs of ribs; narrow thoracic cage; hypoplasia of ischiopubis and flared ilia; lack of ossification of calcaneal facilities; markedly delayed ossification of proximal phalanges and middle phalanges with well-ossified distal phalanges. Ocular hypertelorism, depressed nasal bridge, midface hypoplasia, micrognathia, multiple giant joint dislocations, talipes equinovarus, polyhydramnios. Low-set ears, helix hypoplasia, and stenotic ear canals, hypotonia, hydrocephalus, and seizures occur less regularly. Respiratory problems and cervical spine instability often lead to dying within the newborn period. However, survival to maturity has been documented in a single case, a girl who subsequently gave delivery to an affected baby. Inheritance is autosomal dominant, with most circumstances representing contemporary gene mutations. The neck is short, the Atelosteogenesis, Type I short-limb skeletal dysplasia, Am J Med Genet thirteen:7, 1982. Note the depressed nasal bridge, flexion contractures at knees, and equinovarus place of ft. B, the radiograph reveals lack of calcification of humerus and hypoplasia of a lot of the skeleton. A, Postmortem photograph of new child showing limb shortening, a flat face, radial deviation of a low-implanted thumb, and equinovarus with a big hole between toes 1 and 2. B, Note the quick limbs, small thorax, bifid distal humeri and rounded iliac bones. F and G, Note, in an adult, the shortening of C2 to C7, flattened our bodies of C3 to C6, shortened radius, abnormally formed carpals, and short metacarpals and phalanges. The designation ` `boomerang dysplasia is usually attributed to Kozlowski et al, who noticed the standard boomerang-like shape of the long tubular bones. Boomerang dysplasia is distinguished from atelosteogenesis on the basis of a more extreme defect in mineralization, with complete absence of ossification in some limb bones and vertebrae. Heterozygous mutations associated with the most extreme phenotypes virtually invariably happen in exons 2-5. These are apparently de novo, but germline or somatic mosaicism has been reported for the milder phenotypes. Histology exhibits disorganized cartilage of the creating lengthy bone and multinucleated chondrocytes in areas of a hypocellular cartilage matrix. Severe prenatal growth retardation secondary to limb and trunk shortening with sparing of head. Large fontanels, full forehead, hypertelorism, markedly depressed nasal bridge with horizontal groove, hypoplastic nasal septum, micrognathia, quick neck with extra skin. Severe micromelia, normally symmetric; irregular position of the limbs, generally formed by a single section; absence of discernible joints; talipes equinovarus. The palms and toes are brief and broad and have shortened fingers and toes with poly- or oligodactyly, syndactyly, and hypoplastic nails. Variable delayed calvarial ossification, relative preservation of the thorax, clavicles, sternum and iliac wings with extreme delay in mineralization of the vertebrae, pubis, metacarpal/tarsals, and long tubular bones of the arms and legs, boomerang-shaped femora. Sillence D, et al: Atelosteogenesis syndromes: A evaluate, with comments on their pathogenesis, Pediatr Radiol 27:388, 1997. Odent S, et al: Unusual fan formed ossification in a feminine fetus with radiological options of boomerang dysplasia, J Med Genet 36:330, 1999. Lu J, et al: Filamin B mutations cause chondrocyte defects in skeletal improvement, Hum Mol Genet sixteen:1661, 2007.
|
